분석 파이프라인
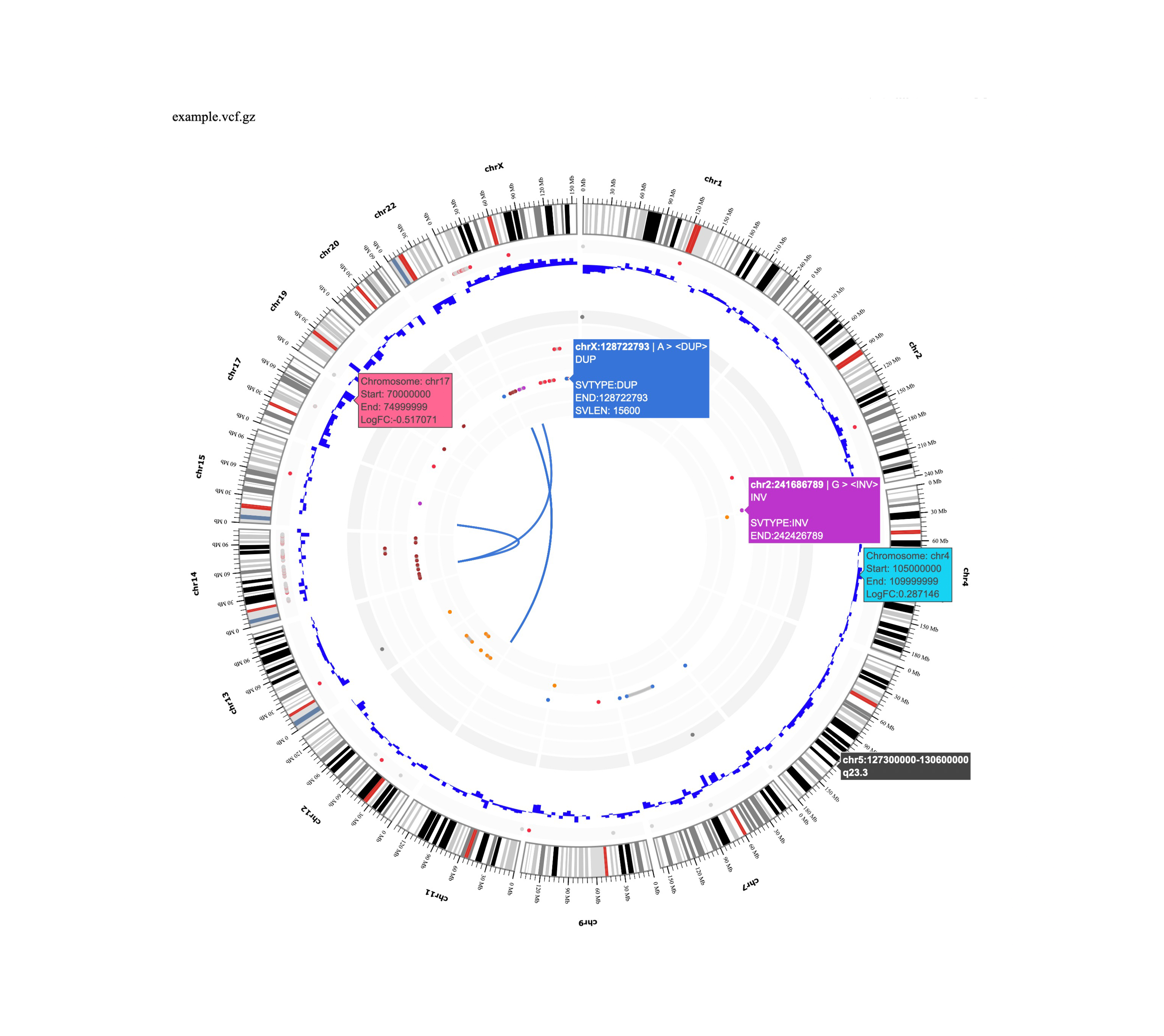
The Whole-genome sequencing(WGS) pipeline is a modular toolkit for processing WGS data. This pipeline takes a FASTQ file as input and provides haplotype call results and annotations and visualizations based on GATK pipeline. First, raw read data with well-calibrated base error estimates in FASTQ format are mapped to the reference genome. The BWA mapping tool is used to align reads to the human genome reference, allowing for up to two mismatches in 30-base seeds, and generate a technology-independent SAM/BAM reference file format. Next, duplicate fragments are marked and removed using Picard(http://picard.sourceforge.net), mapping quality is assessed and low-quality mapped reads are filtered, and Paired-read information is also evaluated to ensure that all mate-pair information is in sync between each read. We then refine the initial alignments with local realignment and identify suspicious regions. Using this information as a covariate along with other technical covariates and known sites of variation, the GATK base quality score recalibration(BQSR) is performed. Germline SNPs and indels are called via local reassembly of haplotypes using the recalibrated and realigned BAM files. Finally, we provide Somalier, a tool to quickly assessing sample relevance from sequencing data in BAM, CRAM or VCF format.
- 카테고리Bioinformatics > Whole-Genome-Sequencing
- 수정일자2025-05-20
 (1).png)
The Single-cell RNA sequencing pipeline is an extensible toolkit for analyzing single-cell gene expression data using the Scanpy framework. It includes methods for preprocessing, visualization, clustering, and differential expression testing. Its Python-based implementation efficiently handles datasets containing more than one million cells. We introduce ANNDATA, a generic class for managing annotated data matrices. The pipeline features: 1. Regression of confounding variables, normalization, and identification of highly variable genes. 2. t-SNE and graph-based (Fruchterman–Reingold) visualizations that show cell-type annotations derived from comparisons with bulk expression data. 3. Clustering of cells and visualization using the Louvain algorithm, with support for other clustering algorithms as well. 4. Ranking differentially expressed genes in clusters to identify marker genes corresponding to bulk expression labels.
- 카테고리Bioinformatics > Single-Cell-RNA-Sequencing
- 수정일자2025-03-26